

ავტორი: რუსუდან პატარქალიშვილი. კავკასიის საერთაშორისო უნივერსიტეტის მე-6 კურსის სტუდენტი.
შესავალი
2015 წელს ალტმანმა და კოლეგებმა აღწერეს LQTS (გახანგრძლივებული QT ინტერვალის სინდრომის) იშვიათი აუტოსომურ-რეცესიული ფორმა, რომელიც ხასიათდება პრეკორდიალურ განხრებში (V1-V4) QT ინტერვალის გარდამავალი/თანმიმდევრული გახანგრძლივებით, T ტალღის ინვერსიებით და ასევე ბავშვთა ასაკში მძიმე და ხშირად რეფრაქტერული ფიზიკური დატვირთვით გამოწვეული პარკუჭოვანი არითმიებით.
გარდა ამისა, მათ აღწერეს მსუბუქი და საშუალო სიმძიმის პროქსიმალური ჩონჩხის მიოპათია, რომელიც მეორადად გამოწვეული იყო TRDN-კოდირებული ტრიადინის ჰომოზიგოტური ან რთული ჰეტეროზიგოტური ვარიანტებით. საინტერესოა, რომ იგივე TRDN ვარიანტებიდან ზოგიერთი იყო ჩართული რეცესიულად მემკვიდრეობით მიღებულ CPVT-ში (კატექოლამინერგული პოლიმორფული პარკუჭოვანი ტაქიკარდია), რაც მიუთითებს, რომ ტრიადინის ნოკაუტის სინდრომი (TKOS) შეიძლება წარმოადგენდეს უნიკალურ კლინიკურ ერთეულს, რომელსაც აქვს როგორც LQTS, ასევე CPVT კლინიკური მახასიათებლები.
მიუხედავად იმისა, რომ TKOS პაციენტებში დაფიქსირებული საშუალო QTc იყო 472 ± მილიწამი, 89%-ში დაფიქსირდა ფიზიკური დატვირთვით გამოწვეული ექტოპია.
გარდა ამისა, TKOS პაციენტების 90%-ს განუვითარდა უეცარი გულის გაჩერების სინდრომი და უეცარი კარდიული სიკვდილი (SCA/SCD), ხოლო მიუხედავად მრავალი სამედიცინო, ქირურგიული (სიმპათექტომია) და აპარატურით თერაპიისა 74%-ს განუვითარდა კარდიული გამწვავებები, რაც ხაზს უსვამს TKOS კლინიკური ფენოტიპის ავთვისებიან ბუნებას. სინდრომის სიმძიმიდან გამომდინარე შეიქმნა რეესტრი, რომლის მეშვეობითაც ხდება კლინიკური მონაცემების შეგროვება, რათა უკეთ იქნას გაგებული ამ პოტენციურად სიცოცხლისთვის საშიში დაავადების ბუნებრივი მიმდინარეობა, კლინიკური ნიშნები და მკურნალობის შედეგები.
შეიქმნა ტრიადინ ნოკაუტის სინდრომის საერთაშორისო რეესტრი, რათა აღიწეროს პაციენტი, რომლსაც გენეტიკურად დადასტურებული ჰომოზიგოტური/რთული ჰეტეროზიგოტური TRDN ნულოვანი მუტაციები აქვთ ან დამახასიათებელი კლინიკური ნიშნები (T ტალღის ინვერსია, QT ინტერვალის გახანგრძლივება, პარკუჭოვანი არითმიები). ტრიადინის ნოკაუტის სინდრომის საერთაშორისო რეესტრი მოიცავს 21 პაციენტს 16 სხვადასხვა ოჯახიდან. აქედან 11 მამრობითი სქესის და 10 მდედრობითი სქესის პაციენტია. პაციენტთა საშუალო ასაკი 18 წელია.
ოცი პაციენტი (95%) წარმოდგენილია გულის გაჩერებით (15, 71%) ან სინკოპეთი (5, 24%) საშუალოდ 3 წლის ასაკში. მსუბუქი ჩონჩხის მიოპათია/პროქსიმალური კუნთების სისუსტე აღინიშნა 6 (29%) პაციენტში. 19 გადარჩენილი პაციენტიდან 16-ს (84%) აღენიშნება T-ტალღის ინვერსიები, ხოლო 10-ს (53%) აღენიშნება QT ინტერვალის გარდამავალი გახანგრძლივება > 480 ms. 9 პაციენტიდან რვას დატვირთვის ტესტის დროს აღენიშნებოდა პარკუჭოვანი ექტოპია. 13 (68%) პაციენტს ჩაედგა იმპლანტირებადი დეფიბრილატორი. მკურნალობის სხვადასხვა სტრატეგიის მიუხედავად, 14 (74%) პაციენტს აღენიშნებოდა კარდიული განმეორებითი გამწვავებები.
ამ ფენოტიპის მქონე პაციენტებს უნდა ჩაუტარდეს TRDN გენეტიკური ტესტირება, რადგან TKOS შეიძლება წარმოადგენდეს გულის აუხსნელი გაჩერების მიზეზი მცირეწლოვან ბავშვებში.
რა არის ტრიადინი? რა როლი აკისრია ტრიადინის ნოკაუტის სინდრომის მიმდინარეობაში?
ტრიადინი წარმოადგენს მნიშვნელოვან ცილას, რომელიც განლაგებულია სარკოპლაზმური ბადის (SR) სპეციალიზებულ უბნებში, სადაც სარკოპლაზმური ბადე ქმნის შეერთებებს სარკოლემასთან. ტრიადინი ქმნის ცილოვან კომპლექსს, რომელიც ასოცირდება გულის რიანოდინური რეცეპტორ 2-ის (RyR2) სარკოპლაზმურ ბადეში Ca2+ გამოთავისუფლების არხებთან. ეს კომპლექსი პასუხისმგებელია კალციუმის აღქმის რეგულაციაზე და გულის კუნთში აღგზნება-შეკუმშვის სწორ კოორდინაციაზე. ტრიადინი დაკავშირებულია რამდენიმე მნიშვნელოვან ცილასთან კალციუმის გამოყოფის ერთეულში, მათ შორის RyR2, Casq2 (კალსეკვესტრინი 2), Jph2 და თამაშობს მნიშვნელოვან როლს როგორც კალციუმის სწორი გამოყოფის რეგულაციაში სარკოპლაზმური ბადიდან RyR2-ის საშუალებით, ისე თავად კალციუმის გამოყოფის ერთეულის სტრუქტურულ სტაბილიზაციაში. სწორედ ტრიადინის ნოკაუტის სინდრომის დროს TRDN გენში ხდება მუტაცია, რაც იწვევს ტრიადინის ცილის დაკარგვას ან დარღვევას, რომელიც ხდება ან ჰომოზიგოტური ან კომპაუნდ ჰეტეროზიგოტური ნულისებური მუტაციების შედეგად, შესაბამისად მისი მუტაციის შემთხვევაში ვითარდება არითმოგენული ფენოტიპი, რაც ნიშნავს რომ სტრუქტურულად გული ნორმალურია, მაგრამ მიდრეკილია პარკუჭოვანი არითმიებისკენ და ფიზიკური დატვირთვის ფონზე გულის უეცარი გაჩერებისკენ.
კერძოდ ტრიადინის მუტაცია იწვევს კალციუმის გამოყოფის ერთეულის სტრუქტურული ბალანსის დარღვევას, L-ტიპის კალციუმის არხისა (LTCC) და RyR2-ის დე-კოლოკალიზაციას, რაც სარკოპლაზმურ ბადეში კალციუმის დაგროვებას და LTCC-ის არაადეკვატურ ინაქტივაციას იწვევს. ეს კი თავის მხრივ განაპირობებს როგორც პარკუჭოვან არითმიებს, ასევე შემცირებულ კუნთოვან ძალას. ამიტომ მიჩნეულია, რომ TKOS-ის მქონე პაციენტებში სარკოპლაზმური ბადის კალციუმის ჭარბი დაგროვება შეიძლება იწვევდეს დაგვიანებულ შემდგომ დეპოლარიზაციებს, მსგავსად CPVT-ისა, ხოლო LTCC-ის დაგვიანებული ინაქტივაცია-გახანგრძლივებულ მოქმედებით პოტენციალს და ადრეულ შემდგომ დეპოლარიზაციებს, ორივე წარმოადგენს ვენტრიკულური არითმიის წინამორბედს.
სტატისტიკა
უეცარი კარდიული სიკვდილი მსოფლიო მასშტაბით საზოგადოებრივი ჯანმრთელობის მნიშვნელოვან ტვირთს წარმოადგენს, რომლის წლიური მაჩვენებელი, სავარაუდოდ, 180 000-დან 450 000-მდე მერყეობს შეერთებულ შტატებში და მსოფლიოში 3.7 მილიონამდე სიკვდილიანობაა. ახალგაზრდებში უეცარ სიკვდილს დამანგრეველი და ღრმა სოციალური გავლენა აქვს, რადგან მხოლოდ შეერთებულ შტატებში ყოველწლიურად 1-დან 35 წლამდე ასაკის დაახლოებით 2000-დან 5000-მდე ახალგაზრდა განიცდის უეცარ კარდიულ სიკვდილს. პოტენციურად ლეტალური გულის არითმიის სინდრომები, როგორიცაა გახანგრძლივებული QT სინდრომი (LQTS), კატექოლამინერგული პოლიმორფული პარკუჭოვანი ტაქიკარდია (CPVT) და იდიოპათიური პარკუჭოვანი ფიბრილაცია, ახალგაზრდებში უეცარი სიკვდილის მნიშვნელოვან ნაწილს წარმოადგენს.
ბოლო დროს, რამდენიმე მცირე კოჰორტული კვლევაში TRDN-კოდირებული გულის ტრიადინი იდენტიფიცირებულია, როგორც ძირითადი მიზეზი ზოგიერთი პაციენტისთვის, რომლებსაც თავდაპირველად LQTS, CPVT ან იდიოპათიური პარკუჭოვანი ფიბრილაციის დიაგნოზი დაუსვეს.
2012 წელს, ტრიადინის ნულოვანი მუტაციები პირველად გამოქვეყნდა, როგორც რეცესიულად მემკვიდრეობითი არითმოგენური დაავადების ძირითადი მიზეზი, როდესაც CPVT დიაგნოზის მქონე 97 პაციენტიდან 2-ს (2%) ქონდა დიაგნოსტირებული, მაგრამ RYR2 და CASQ2 გენებზე გენოტიპურად უარყოფითები იყვნენ, აღმოაჩნდათ ან ჰომოზიგოტური ან რთული ჰეტეროზიგოტური ტრიადინის ნულოვანი მუტაციები. მომდევნო სამი კლინიკური შემთხვევის აღწერაშიც დაფიქსირდა ტრიადინის ნულოვანი მუტაციები იმ პაციენტებში, რომელთაც თავდაპირველად CPVT ან იდიოპათიური ვენტრიკულური ფიბრილაცია ქონდათ დიაგნოსტირებული.
TKOS-ის შემთხვევების რაოდენობა გაცილებით მაღალი შეიძლება იყოს იმ რეგიონებში, სადაც მოსახლეობის უმრავლესობა შავკანიანია-მაგალითად, ცენტრალურ აფრიკაში. გარდა ამისა, TKOS რეცესიულად მემკვიდრეობით გადაცემადი დაავადებაა, რაც ნიშნავს, რომ შემთხვევების სიხშირე შესაძლოა მაღალი იყოს იმ რეგიონებში, სადაც ნათესაური ქორწინებები ხშირია-კერძოდ, შუა აღმოსავლეთში, სამხრეთ აზიასა და ჩრდილოეთ აფრიკაში, სადაც ქორწინებების 20%-დან 50%-მდე შეიძლება ნათესაურ კავშირში მყოფ პირებს შორის სრულდებოდეს. საინტერესოა, რომ საერთაშორისო Triadin Knockout Syndrome-ის რეესტრში დარეგისტრირებულ 16 ოჯახიდან 6 ინდური ან არაბული წარმოშობისაა, და ამ ოჯახებიდან 2-ში დადასტურებულია, რომ მშობლები პირველი ან მეორე რიგის ნათესავები არიან.
ბოლოდროინდელმა მონაცემებმა, რომლებიც მიღებულია საერთაშორისო TKOS რეესტრიდან, აჩვენა, რომ TKOS-ის მქონე პაციენტების 95%-ს აღენიშნება სიმპტომები და დიაგნოზი ხშირად დგინდება საშუალოდ 3 წლის ასაკში, ხოლო ყველა შემთხვევა ვლინდება 10 წლამდე. გარდა ამისა, პაციენტების 81%-ს გადაუტანია მინიმუმ ერთი გულის გაჩერება, ხოლო 10%-ს ჰქონდა უეცარი კარდიული სიკვდილი. TRDN გენის მხოლოდ ერთ ნულოვან ალელზე ჰეტეროზიგოტი პირები დაავადებას არ ავლენენ.
ისტორია
2015 წელს, ალტმანმა და მისმა კოლეგებმა აღმოაჩინეს, რომ TRDN გენის ჰომოზიგოტური ან კომპაუნდ-ჰეტეროზიგოტური ფრემშიფტ მუტაციები იყო დაავადების მიზეზი იმ ხუთ პაციენტში, რომელთაც თავდაპირველად გენეტიკურად გაურკვეველი LQTS (გახანგრძლივებული QT ინტერვალის სინდრომი) ჰქონდათ დიაგნოსტირებული. ამ პაციენტებს საერთო ელექტროკარდიოგრაფიული და კლინიკური ფენოტიპი ჰქონდათ, რომელიც შეიცავდა როგორც LQTS-ის, ისე CPVT-ის არატიპურ ნიშნებს. შედეგად, ტერმინი ‘triadin knockout სინდრომი’ (TKOS) შემოიტანეს. აღსანიშნავია, რო ხუთივე პაციენტმა აჩვენა ან მუდმივი, ან დროებითი QT-ის გახანგრძლივება, აღენიშნებოდათ მკვეთრი T ტალღის ინვერსიები პრეკორდიულ განხრებში V1-დან V4-მდე და დაავადების მძიმედ გამოხატული კლინიკური მიმდინარეობა-ადრეული ბავშვობის პერიოდში ფიზიკური დატვირთვით გამოწვეული მოულოდნელი გულის გაჩერების (SCA) ეპიზოდებით. უმეტეს მათგანს მკურნალობის მიუხედავად კვლავ განმეორებული კარდიული გართულებების ეპიზოდები აღენიშნებოდა. გარდა ამისა, ხუთიდან ორ პაციენტს აღენიშნებოდა მსუბუქი ჩონჩხის კუნთების სისუსტეც. აღსანიშნავია, რომ ტრიადინის mRNA-ს ალტერნატიული სპლაისინგი იწვევს ქსოვილზე დამოკიდებული ტრიადინის იზოფორმების წარმოქმნას, რომლებიც გვხვდება როგორც გულის, ისე ჩონჩხის კუნთებში. იმის გამო, რომ ეს სხვადასხვა იზოფორმები ერთნაირია ამინომჟავათა 264-ე პოზიციამდე, დღემდე აღმოჩენილი ყველა მუტაცია, რომელიც იწვევს ტრიადინის არარსებობას გულში (cardiac triadin null), ასევე იწვევს მის არარსებობას ჩონჩხის კუნთებშიც (skeletal muscle triadin null). ჯერჯერობით უცნობია, რატომ არ აღენიშნება უფრო მეტ TKOS პაციენტს ჩონჩხის კუნთების დაზიანება, თუმცა შესაძლებელია, ნაკლები ყურადღება ექცევა ამ სიმპტომებს, რადგან ფენოტიპი სუსტად არის გამოხატული, და ამ პაციენტების უმეტესობას არ გაუვლია შეფასება ნევროკუნთოვანი სპეციალისტის მიერ.
ამ სინდრომის პოტენციურად ლეტალური ბუნების გათვალისწინებით, გადაწყდა, უკეთ გაეგოთ TKOS-ით დაავადებული პაციენტების ფენოტიპი და მკურნალობის შედეგები, საერთაშორისო ტრიადინის ნოკაუტის სინდრომის რეესტრის (ITKOSR) შექმნით. საერთო ჯამში ამ რეესტრით 21 პაციენტია დაფიქსირებული, 16 განსხვავებული ოჯახიდან, რომლებიც აკმაყოფილებდნენ ITKOSR-ში ჩართვის კრიტერიუმებს.
21 პაციენტიდან 14 ადრე იყო აღწერილი ლიტერატურაში. საერთო ჯამში, 11 (52%) მამაკაცი და 10 (48%) ქალი იყო. რეესტრში რეგისტრაციის საშუალო ასაკი იყო 18±14 წელი (3-დან 45 წლამდე). პაციენტები იყვნენ სხვადასხვა ეთნიკური წარმომავლობის, მათ შორის თეთრკანიანები (8/21, 38%), ინდოელები (4, 19%), ლათინოამერიკელები (3, 14%), აფრიკელები (2, 9%) და არაბები (2, 9%), ასევე 2 (9%) პაციენტი, რომლებიც ნახევრად თეთრკანიანები და ნახევრად აფრიკელები იყვნენ. TKOS დიაგნოზამდე პაციენტებს დაუსვეს შემდეგი დიაგნოზი:LQTS (8/21, 38%), CPVT (7, 33%), იდიოპათიური პარკუჭოვანი ფიბრილაცია (4, 19%), ფიზიკური დატვირთვით გამოწვეული სინკოპე (1, 5%), ან აუხსნელი უეცარი კარდიული სიკვდილი (1, 5%).
გენეტიკა
ITKOSR კვლევაში ჩართული ყველა პაციენტი ატარებდა TRDN გენის ან ჰომოზიგოტურ (12 პაციენტი 21-დან, 57%) ან კომპაუნდ ჰეტეროზიგოტურ (9 პაციენტი, 43%) ნულოვან მუტაციებს. ჯამში გამოვლინდა TRDN გენის 13 უნიკალური პათოგენური ან სავარაუდოდ პათოგენური ვარიანტი. ამ ვარიანტთაგან 7 ადრე იყო აღწერილი TKOS-ის კლინიკურ შემთხვევებში, ხოლო დარჩენილი 6 მუტაცია პირველად აღწერილია სწორედ ამ კვლევის ფარგლებში. მეტწილად, აღმოჩენილი მუტაციები წარმოადგენდა: ფრემშიფტ მუტაციებს (6 ვარიანტი, 46%), ნონსენს მუტაციებს (2 ვარიანტი, 15%), ან სპლაისინგის პროცესზე მოქმედ მუტაციებს (2 ვარიანტი, 15%), რომლებიც იწვევენ ნაადრევი ტერმინაციის კოდონის წარმოქმნას და შედეგად-ცილის ნონსენს-მედიირებულ დეგრადაციას. ოთხი პათოგენური ვარიანტი გამოვლენილია სხვადასხვა, ურთიერთდაუკავშირდებელ ოჯახებში, რაც მათ რეცესიულ მემკვიდრეობითობას კიდევ უფრო ადასტურებს-p.D18fs*13 (2 ოჯახი), p.N9fs*5 (2 ოჯახი), p.K147fs*0 (5 ოჯახი) და p.Q205* (2 ოჯახი). დამატებით, გამოვლინდა:ორი (15%) პათოგენური მისსენს მუტაცია (p.T59R და p.T59M) და ერთი (8%) დელეცია, რომელიც მოიცავს ექსონ 2-ს.
მისსენს მუტაცია p.T59R, რომელიც ლოკალიზებულია ტრიადინის ტრანსმემბრანულ დომენში, ადრეულ კვლევებში უკვე დახასიათებულია როგორც მუტაცია, რომელიც იწვევს არასტაბილური ცილის წარმოქმნას და მის პროტეასომულ დეგრადაციას. სავარაუდოდ, იმავე ამინომჟავურ ნაშთზე მომხდარი p.T59M მუტაციაც იწვევს ანალოგიურ შედეგს- არასტაბილური ცილის წარმოქმნას, რომელიც საბოლოოდ ფუნქციურად ნულოვან ალელად ითვლება. ჰომოზიგოტური დელეცია TRDN გენის ექსონ 2-ის, რომელიც კოდირებს ტრიადინის N-ტერმინალურ უბანსა და მთელ ტრანსმემბრანულ დომენს, გამოვლინდა ერთ პაციენტში და პირველად აღწერეს O’Callaghan და კოლეგებმა.
სიმპტომები
საერთო ჯამში, 21 პაციენტიდან 20 (95%) სიმპტომური იყო და სულ მცირე ერთი კარდიოლოგიური შემთხვევა ჰქონდათ გადატანილი. იმ პაციენტთაგან, ვისაც პირველი კარდიული შემთხვევის დრო დაფიქსირებული ჰქონდა (18 პაციენტი 86%), პირველი შემთხვევა ადრეულ ასაკში გამოვლინდა-საშუალოდ 3.2 წლის ასაკში. დამატებით, სიმპტომური პაციენტების 83% დაავადება 5 წლის ასაკამდე გამოუვლინდა, ხოლო ყველა შემთხვევა-10 წლამდე. გულისწასვლა (სინკოპე) იყო პირველი სიმპტომი 5 პაციენტში (24%), მაშინ როდესაც 15 პაციენტი (71%) პირველად გამოვლინდა გულის უეცარი გაჩერებით (SCA). აღსანიშნავია, რომ სინკოპით დებიუტირებული 5-დან 2 პაციენტს შემდგომში განუვითარდა SCA, რაც იმას ნიშნავს, რომ 21-დან 17 პაციენტს (81%) საბოლოოდ აღენიშნებოდა SCA. ყველაზე გავრცელებული გამომწვევი ფაქტორი იყო ფიზიკური დატვირთვა, რომელიც ასოცირდებოდა სულ მცირე ერთი კარდიული ეპიზოდით 21-დან 17 პაციენტში (81%). სხვა გამომწვევი ფაქტორები მოიცავდა ხმოვან სტიმულებს, შიშს და ძილს, თუმცა ბევრი ეპიზოდი კონკრეტულ გამომწვევთან არ იყო ასოცირებული. გარდა მძიმე კარდიული სიმპტომებისა, 6 პაციენტს (29%) აღენიშნებოდა მსუბუქი ჩონჩხის მიოპათია ან მცირე პროქსიმალური კუნთოვანი სისუსტე. ამჟამად 19 პაციენტი ცოცხალია, ხოლო 2 პაციენტი გარდაიცვალა კარდიული ეპიზოდების შემდეგ.
კლინიკური შეფასება
ყველა ცოცხლად დარჩენილ 19 TKOS პაციენტს ჩაუტარდა მინიმუმ ერთი სტანდარტული 12-განხრიანი ელექტროკარდიოგრამა. მათგან 10 პაციენტს (53%) დაუფიქსირდა QT ინტერვალის გახანგრძლივება (QTc >480 მილიწამი) სულ მცირე ერთ ეკგ-ზე. მიუხედავად იმისა, რომ QT-ის გახანგრძლივება დროებითი იყო 10-დან 9 პაციენტში, ერთ შემთხვევაში QTc გახანგრძლივება იყო მუდმივი ხასიათის. დამატებით, ცოცხლად დარჩენილი 19 პაციენტიდან 16-ს (84%) აღენიშნება T ტალღის ინვერსიები (TWI), საიდანაც 13 პაციენტს (68%) ინვერსიები აღენიშნება გულმკერდის (პრეკორდიალურ) განხრებზე, ძირითადად V1-V3 ან V1-V4. მიუხედავად იმისა, რომ T ტალღების ინვერსია ბავშვებში შესაძლოა ნორმალური იყოს, მათი თანმიმდევრული გაგრძელება V3 ელექტროდიდან V4-სა და განსაკუთრებით V5-მდე ითვლება პათოლოგიური ეკგ ფენოტიპის ნიშნად. იქნება თუ არა TWI შენარჩუნებული სრულწლოვანებაშიც-ეს ჯერ უცნობია, თუმცა TKOS-ის სხვა კლინიკურ ნიშნებთან ერთად, TWI შესაძლოა გახდეს მნიშვნელოვანი დიაგნოსტიკური კრიტერიუმი, რომელიც TKOS-ს განასხვავებს სხვა არითმიული სინდრომებისგან.
ყველა 16 პაციენტს, ვისაც ჩაუტარდა ექოკარდიოგრაფია, დაუფიქსირდა სტრუქტურულად ნორმალური გული, რაც გულის შენების ნორმალურობას ადასტურებს. დატვირთვის ტესტი ჩაუტარდა 9 პაციენტს, რომელთაგან 8 პაციენტს (89%) გამოუვლინდა ვენტრიკულური ექტოპია. დატვირთვით გამოწვეული ექტოპიური აქტივობა ვლინდებოდა შემდეგი სახით: იზოლირებული ნაადრევი ვენტრიკულური კომპლექსები (PVCs), ბიგემინია, კუპლეტები და ტრიპლეტები. აღსანიშნავია, რომ არცერთ პაციენტს არ დაუფიქსირდა არამუდმივი ვენტრიკულური ტაქიკარდია (nonsustained VT) ან ბიდირექციული ვენტრიკულური ტაქიკარდია (bidirectional VT).
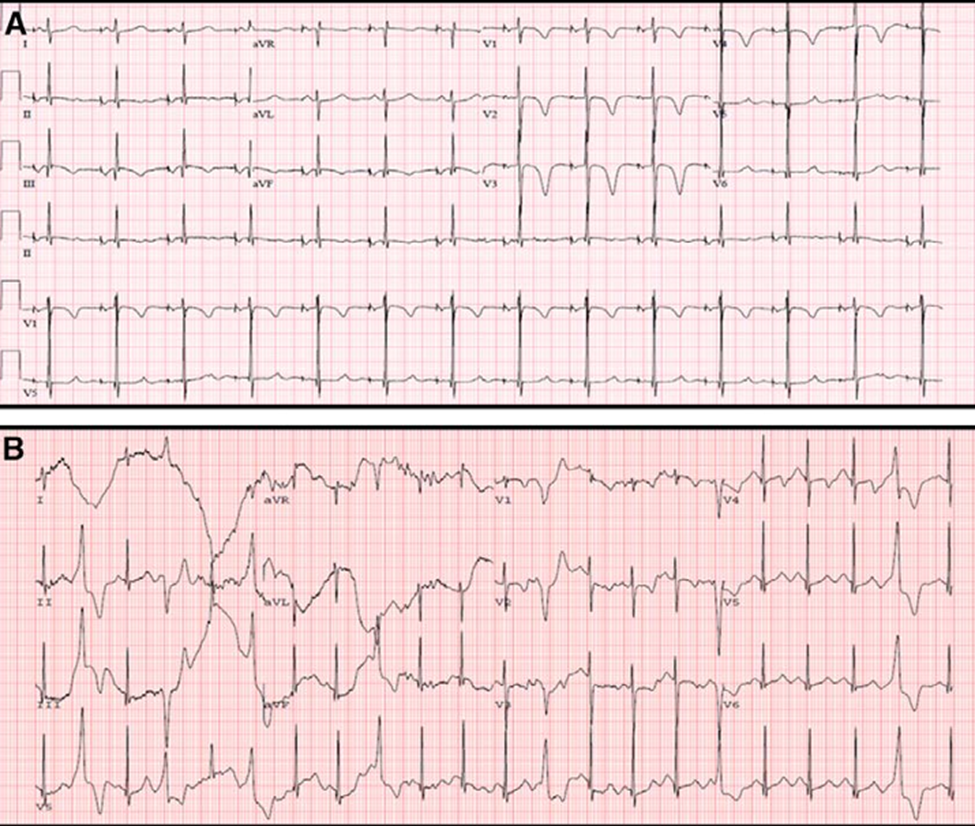
აღნიშნულ ეკგ-ზე ასახულია ტრიადინის ნოკაუტის სინდრომის ელექტროკარდიოგრაფიული დამახასიათებელი ნიშნები. A-მოსვენების 12-განხრიანი ელექტროკარდიოგრამა, რომელიც აჩვენებს T-ტალღის ინვერსიებს პრეკორდიალურ განხრაში V1-V4. B-12-განხრიანი ელექტროკარდიოგრამა დატვირთვის ტესტიდან, რომელიც აჩვენებს პარკუჭების ხშირ ექტოპიურ აქტივობას.
საინტერესოა ისიც, რომ TKOS-ის პაციენტებს, რომლებმაც განიცადეს BCE, ჰქონდათ სტატისტიკურად მნიშვნელოვნად უფრო გახანგრძლივებული QTc, ვიდრე მათ, ვისაც მკურნალობის ფონზე BCE არ ჰქონია. შეიძლება თუ არა QTc ინტერვალი გახდეს პროგნოსტიკური მაჩვენებელი იმ პაციენტებისთვის, რომლებსაც აქვთ BCE-ს განვითარების ყველაზე მაღალი რისკი-ჯერ არ არის საბოლოოდ დადგენილი. აღრიცხული პაციენტების ჯგუფი საშუალოდ QTc 472 მილიწამით გამოირჩევა, რაც მსგავსია LQT1-3 ტიპის მქონე პაციენტების წარსულ კვლევებში დაფიქსირებულ მაჩვენებლებთან (472 მმწ-ს მსგავსად) და მნიშვნელოვნად უფრო გრძელია, ვიდრე CPVT-ით (კატექოლამინერგული პოლიმორფული ვენტრიკულური ტაქიკარდია) პაციენტებში დაფიქსირებული QT ინტერვალი. საპირისპიროდ, TKOS-ის პაციენტების 89%-ს სტრეს-ტესტირებისას ვარჯიშის პიკზე აღენიშნება გამოწვეული ექტოპიური უკმარისობა-რაც CPVT-ის დამახასიათებელი ნიშანია და იშვიათად, გვხვდება LQTS-ის მქონე პაციენტებში.
ერთ-ერთი დამახასიათებელი ნიშანი ტრიადინის ნოკაუტის სინდრომისთვის არის გახანგრძლივებული QT სინდრომი, რომლის სამი ტიპი არსებობს:LQT1, LQT2, LQT3. გახანგრძლივებული QT სინდრომი (LQTS) არის პარკუჭოვანი მიოკარდიუმის რეპოლარიზაციის დარღვევა, რომელიც ხასიათდება ელექტროკარდიოგრამაზე QT ინტერვალის გახანგრძლივებით, რამაც შეიძლება გამოიწვიოს სიმპტომური პარკუჭოვანი არითმიები და უეცარი კარდიული სიკვდილის (SCD) გაზრდილი რისკი. LQTS-ით დაავადებულ პაციენტებში ძირითადი სიმპტომებია სინკოპე, კრუნჩხვები, SCD. LQTS ასოცირდება სიცოცხლისთვის საშიში გულის არითმიის, რომელიც ცნობილია როგორც torsades de pointes (TdP) გაზრდილი რისკის მქონე. LQTS-ის ხშირად დაფიქსირებული მახასიათებლების ასახსნელად ორი წამყვანი პათოფიზიოლოგიური ჰიპოთეზა გაჩნდა:
1)ფართო და მზარდი კლინიკური და გენეტიკური მტკიცებულებები ადასტურებს გულის იონური არხების დარღვევების მნიშვნელობას, რაც იწვევს მოქმედების პოტენციალის გახანგრძლივებას. ამ მონაცემებზე დაყრდნობით, თანდაყოლილი LQTS იონური არხების დაავადებად ითვლება და ყველაზე გავრცელებულებულად.
2)დაკვირვებამ, რომ მემკვიდრეობითი ფორმის დროს TdP დაუყოვნებელი გამომწვევი მიზეზი ხშირად სიმპათიკური ტონუსის უეცარი მატებაა (რაც შეძენილ ფორმაში არ შეინიშნება), გამოიწვია ჰიპოთეზა, რომ თანდაყოლილი LQTS შეიძლება გამოწვეული იყოს გულის სიმპათიკური ინერვაციის დისბალანსით.
LQTS-ის მქონე პირთა უმრავლესობას დიაგნოზის დასმისას სიმპტომები არ აღენიშნება და მთელი ცხოვრება ასე რჩება. სიმპტომურ პაციენტებს ყველაზე ხშირად აღენიშნებათ LQTS-ით გამოწვეული სინკოპე ან სინკოპე, რასაც მოჰყვება გენერალიზებული კრუნჩხვები. სინამდვილეში, LQTS-ის მქონე ბევრ პაციენტს კვლავ არასწორად უსვამენ ეპილეფსიის დიაგნოზს და არასწორად მკურნალობენ ანტიეპილეფსიური მედიკამენტებით. თანდაყოლილი LQTS-ის მქონე პაციენტთა მნიშვნელოვან უმცირესობას დაავადების პირველი გამოვლინების სახით უეცარი კარდული სიკვდილი აღენიშნება. LQTS დამახასიათებელია ოჯახური ისტორია, ასევე შემდეგიდან ერთ-ერთი:აუხსნელი სინკოპე, კრუნჩხვები ან გულის გაჩერება, რომელსაც წინ უძღოდა ფიზიკური დატვირთვა ან ემოცია. როცა ვითარდება VT-თან დაკავშირებულ სინკოპალურ ეპიზოდები შეიძლება თან ახლდეს ტონური-კლონური მოძრაობები და შესაძლოა არასწორად დიაგნოზირებული იყოს, როგორც პირველადი კრუნჩხვითი აშლილობა. პირველადი კრუნჩხვითი აშლილობისა და VT გამოწვეული გენერალიზებული კრუნჩხვების დიფერენცირება შეიძლება რთული იყოს და შესაძლოა, ეს ერთეულები ერთმანეთს გადაფარავდეს. გარდა ამისა, პაციენტებს, რომლებსაც აღენიშნებათ ეპილეფსია და ბოლო დროს გადატანილი კრუნჩხვა (წინა ორი წლის განმავლობაში), ასევე პაციენტებს, რომლებიც იღებენ ანტიეპილეფსიურ მედიკამენტებს ნატრიუმის არხის ბლოკატორების თვისებებით (მაგ. ფენიტოინი, კარბამაზეპინი, გაბაპენტინი და ა.შ.), როგორც ჩანს, აქვთ სტრესული კარდიომიოპათიის მომატებული რისკი.
გენეტიკური ტესტირება
კლინიკური გენეტიკური ტესტირება წარმოადგენს სტანდარტულ მკურნალობას LQTS-ის გამომწვევი ვარიანტების იდენტიფიცირებისთვის ნებისმიერ პაციენტში, რომლისთვისაც განიხილება LQTS-ის კლინიკური დიაგნოზი. მაიოს კლინიკის 541 პაციენტის სერიაში გენეტიკური ტესტირების საერთო შედეგი დაახლოებით 50 პროცენტი იყო და კორელაციაში იყო დაავადების სიმძიმის კლინიკურ მაჩვენებლებთან:
1)პათოგენური ვარიანტის იდენტიფიცირების ალბათობა პროგრესულად იზრდებოდა QTc-ის ზრდასთან ერთად, მერყეობდა 0-დან 62 პროცენტამდე, როდესაც QTc იზრდებოდა ყველაზე დაბალი (<400 ms) ყველაზე მაღალ (>480 ms) კატეგორიამდე.
2)კლინიკური LQTS დიაგნოსტიკური ინსტრუმენტი, რომელიც პროგნოზირებს LQTS-ის ალბათობას, შვარცის LQTS ქულა, გამოიყოფა QTc-დან, სიმპტომებიდან და ოჯახური ისტორიიდან. პაციენტებში, რომლებსაც აქვთ შვარცის LQTS ქულა >3.5, რაც მიუთითებს LQTS-ის მაღალ ალბათობაზე, დაავადების გამომწვევი ვარიანტი უფრო ხშირად გამოვლინდა, ვიდრე მათ, ვისაც ჰქონდათ <3.5 ქულა.
დიფეენციალური დიაგნოზი
1)ანდერსენ-ტავილის სინდრომი-ანუ ჰიპოკალიემიური პერიოდული დამბლა გულის არითმიით, იშვიათი აუტოსომურ-დომინანტური დაავადებაა, რომელიც ხასიათდება დამბლის ეპიზოდებით, პარკუჭოვანი არითმიებით და დისმორფული ნიშნებით. აღინიშნება QTU ინტერვალის გახანგრძლივება. ეკგ-ზე გამოვლინდა შემდეგი ანომალიები: გახანგრძლივებული ტერმინალური T ტალღის დაღმავალი დახრილობა, ორფაზიანი U ტალღები კიდურების განხრებში, ფართო T-U შეერთება, განსხვავებით ორფაზიანი T ტალღებისგან LQT2 ინტერვალში, გადიდებული U ტალღები, რომლებიც წარმოიქმნება T ტალღის დასრულების შემდეგ განსხვავებული იზოელექტრული სეგმენტით.
2)ტიმოთეს სინდრომი-იშვიათი ჩვილების შემთხვევაა, რომლებსაც აღენიშნებათ QT ინტერვალის მძიმე გახანგრძლივება და სინდაქტილია, რომელიც მოიცავს როგორც თითებს, ასევე ფეხის თითებს, ხშირად თან ახლავს ღია არტერიული სადინარი და სხვა გულის ანომალიები. LQTS-ის ოჯახური ანამნეზის არარსებობა იმაზე მიუთითებს, რომ ეს დარღვევა სპონტანური de novo ვარიანტების შედეგია. მიუხედავად იმისა, რომ ტიმოთეს სინდრომი უკიდურესად იშვიათია, შესაძლოა, გონივრული იყოს სკრინინგის ელექტროკარდიოგრამის ჩატარება QT ინტერვალის შესაფასებლად კომპლექსური სინდაქტილიის მქონე ბავშვებში.
3)SIDS, SUDY-კვლევებმა აჩვენა, რომ SIDS-ის დაახლოებით 5%-10% და SUDY-ის დაახლოებით 25%-30% შეიძლება გამოწვეული იყოს მემკვიდრეობითი გულის დაავადებებისადმი მგრძნობელობის გენებში არსებული ვარიანტებით-განსაკუთრებით იმით, რაც დაკავშირებულია ჩენელოპათიებსა და კარდიომიოპათიებთან. უეცარი ჩვილის სიკვდილის სინდრომის (SIDS) შემთხვევების დაახლოებით 5-10%, ასევე აუხსნელი საშვილოსნოსშიდა ნაყოფის სიკვდილის ზოგიერთი შემთხვევა, შეიძლება გამოწვეული იყოს LQTS, თუმცა SIDS-ის დაბალი შემთხვევები ართულებს თანდაყოლილ LQTS-ზე, როგორც პოტენციურ ეტიოლოგიაზე ზუსტი გავლენის დადგენას.
მკურნალობა
ყველა 19 გადარჩენილი პაციენტი მკურნალობს ბეტა-ბლოკერებით. აბსოლუტური უმრავლესობა ამჟამად იღებს ან ნადოლოლს (14 პაციენტი, 74%) ან პროპრანოლოლს (4 პაციენტი, 21%), ხოლო თითო პაციენტს (5%) ენიშნა ბისოპროლოლი და მეტოპროლოლი. ბეტა-ბლოკერებით მკურნალობასთან ერთად, 6 პაციენტს (32%) დამატებით დაენიშნა ნატრიუმის არხის ბლოკატორი (ფლეკაინიდი-5 პაციენტი, მექსილეტინი-1 პაციენტი) ან კალციუმის არხის ბლოკატორი (ვერაპამილი-1 პაციენტი). მარცხენა გულის სიმპათიკური დენერვაცია (LCSD) ჩაუტარდა 9 პაციენტს (47%), ხოლო 3 მათგანმა (15%) შემდგომში გაიკეთა მარჯვენა გულის სიმპათიკური დენერვაციაც. სულ 14 პაციენტს (74%) ჩაუტარდა იმპლანტირებადი მოწყობილობის ჩანერგვა. 13 პაციენტს (68%) ჩაუნერგეს იმპლანტირებადი კარდიოვერტერ-დეფიბრილატორი (ICD), ხოლო 1 პაციენტს (5%) ჩაუყენდა მუდმივი პეისმეიკერი ბეტა-ბლოკერის დოზის გაზრდის შესაძლებლობისთვის. ბეტა-ბლოკერები, რომლებიც საკმაოდ ეფექტურია LQTS-ისა და CPVT-ის პაციენტების მკურნალობაში, ვერ ახერხებენ გულის არითმიული მოვლენების (BCE) პრევენციას TKOS-ის პაციენტების 71%-ში.
მიუხედავად იმისა, რომ ზოგიერთ პაციენტს ბეტა-ბლოკერისა და ფლეკაინიდის კომბინირებული თერაპიის ფონზე არ განუვითარებია კლინიკური მოვლენები-რაც კლინიკურად და ექსპერიმენტულად დამტკიცებულია, რომ აუმჯობესებს არითმიების კონტროლს CPVT-ის დროს-სხვა პაციენტებს, რომლებსაც მკურნალობად ფლეკაინიდი ან მექსილეტინი მიეღოთ, მაინც განუვითარდათ მრავალჯერადი BCE.
მიუხედავად სხვადასხვა მკურნალობის სტრატეგიებისა, 19 გადარჩენილი TKOS პაციენტიდან 14-ს (74%) აღენიშნა თერაპიის ფონზე განვითარებული BCEs (გულის არითმიული მოვლენები). სინამდვილეში, ამ პაციენტებიდან მხოლოდ ერთს ჰქონდა მხოლოდ ერთი BCE, ხოლო 5 პაციენტს (26%) აღენიშნა 5-ზე მეტი BCE. TKOS დიაგნოზის მქონე პაციენტების უმრავლესობას არაერთგზის განუვითარდა BCEs, თანაც სხვადასხვა მკურნალობის კომბინაციის დროს. გადარჩენილი პაციენტების უმეტესობა (17 პაციენტი, 89%) თავდაპირველად მკურნალობდა მხოლოდ ბეტა-ბლოკერებით ან ბეტა-ბლოკერებისა და ნატრიუმის ან კალციუმის არხის ბლოკატორების კომბინაციით. მხოლოდ მედიკამენტური თერაპიის პირობებში, ამ 17 პაციენტიდან 12-ს (71%) განუვითარდა მინიმუმ ერთი BCE თერაპიის დროს და სხვებს 1-დან 10-მდე ან მეტი BCE. ვინაიდან BCE სიხშირე ბევრი იყო მედიკამენტური მკურნალობის მიუხედავად, შეიძლება ვიმსჯელოთ გარკვეული შემთხვევებში მკურნალობისადმი დაუმორჩილებლობაზე, მიუხედავად იმისა რომ პაციენტების უმრავლესობა მკაცრად იცავდა დანიშნულ ფარმაკოლოგიურ მკურნალობას.
BCE-ის გადატანის შემდეგ, 6 პაციენტს ჩაუტარდა მარცხენა გულის სიმპათიკური დენერვაცია (LCSD). დამატებით, 2 პაციენტი თავდაპირველად მკურნალობა მედიკამენტოზურად და მათთან ერთადვე ჩატარდა LCSD. მიუხედავად მედიკამენტური თერაპიისა და LCSD-ის ჩატარებისა, ამ 8-ვე პაციენტს (100%) განუვითარდა BCE (თითოეულს 1-დან 3-მდე შემთხვევა). ორი პაციენტი, რომლებიც მკურნალობდნენ მედიკამენტურად და ჩაიტარეს LCSD, შემდგომში გადავიდნენ მარჯვენა გულის სიმპათიკურ დენერვაციაზეც. კიდევ ერთმა პაციენტმა მაშინვე, მედიკამენტური მკურნალობიდან, პირდაპირ ჩაიტარა ორმხრივი სიმპათიკური დენერვაცია. ამ დროისთვის, ამ სამიდან ერთმა პაციენტმა (33%), რომელიც მკურნალობდა მედიკამენტურად + LCSD + მარჯვენა დენერვაციით, კვლავ განიცადა BCE. ასევე აღსანიშნავია, რომ 12 განხრიანი ელექტროკარდიოგრამის მიხედვით, პაციენტებს, რომლებსაც განუვითარდათ BCE, საშუალოდ აქვთ მნიშვნელოვნად გახანგრძლივებული QTc ინტერვალი (487±25 ms), შედარებით მათთან, ვისაც BCE არ ჰქონია (429±15 ms).
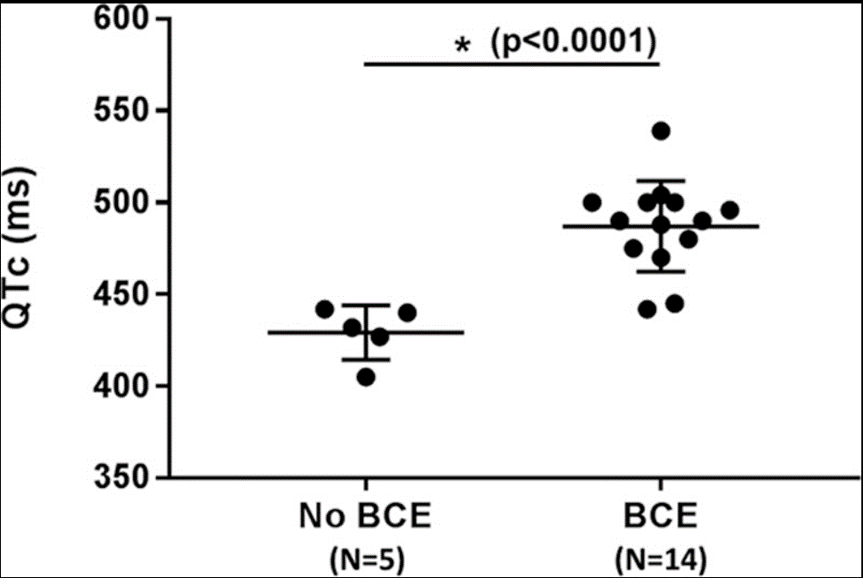
სურათზე გამოსახულია კორექტირებული QT ინტერვალის (QTc) შედარება ტრიადინის ნოკაუტის სინდრომის (TKOS) მქონე პაციენტებში, რომლებმაც განიცადეს ან არ განუცდიათ არითმიული გამწვავება (BCE). პაციენტები, რომლებმაც განიცადეს არითმიული გამწვავება (BCE) ან არ განუცდიათ (No BCE), შედარებულნი იყვნენ QTc ინტერვალის (მილისეკუნდებში) მიხედვით. BCE-ს მქონე პაციენტებს ჰქონდათ მნიშვნელოვნად გახანგრძლივებული QTc (487±25 ms, n=14), შედარებით იმ პაციენტებთან, რომლებსაც BCE არ განუცდიათ (429±15 ms, n=5). თითოეული წერტილი გრაფიკზე წარმოადგენს ერთ პაციენტს. მონაცემები წარმოდგენილია საშუალო მნიშვნელობით ± სტანდარტული გადახრით.
ამჟამად ერთი პაციენტი იღებს კალციუმის არხის ბლოკატორ ვერაპამილს, ბეტა-ბლოკერთან და ფლეკაინიდთან ერთად. მიუხედავად იმისა, რომ ერთი პაციენტის მონაცემებით ძნელია რაიმე დასკვნის გაკეთება კალციუმის არხის ბლოკატორების თერაპიული პოტენციალის შესახებ TKOS-ში, ასეთი პრეპარატები შესაძლოა წარმოადგენდეს თეორიულ ალტერნატივას, განსაკუთრებით თუ გავითვალისწინებთ დაავადების მექანიზმს-L-ტიპის კალციუმის არხის (LTCC) ინაქტივაციის დაყოვნება. თუმცა, ვერაპამილისა და ნიფედიპინის ეფექტიანობა შესაძლოა შეზღუდული იყოს, რადგან ცნობილია, რომ ისინი მიზნად ისახავენ კალციუმის პიკურ დენსაც, ხოლო არა LTCC-ის ინაქტივაციის პროცესს, რაც TKOS-ის მექანიზმში უფრო გადამწყვეტ როლს თამაშობს.
მიუხედავად იმისა, რომ მარცხენა გულის სიმპათიკური დენერვაცია (LCSD) ცნობილია, როგორც ეფექტური მკურნალობის მეთოდი სხვადასხვა არითმიული სინდრომის მქონე პაციენტებისთვის, ყველა TKOS პაციენტს, ვისაც ჩაუტარდა LCSD, მაინც განუვითარდა მინიმუმ ერთი BCE (არითმიული მოვლენა) LCSD-ის შემდეგ. თუმცა, BCE-ის სიხშირე ამ პაციენტებში შედარებით შემცირდა მხოლოდ მედიკამენტურ თერაპიასთან შედარებით. მიუხედავად იმისა, რომ მარჯვენა გულის სიმპათიკური დენერვაციის დამატება შესაძლოა იყოს ეფექტური სტრატეგია TKOS-ის ზოგიერთი პაციენტისთვის, ამ დროისთვის არსებული მტკიცებულება ჯერ კიდევ არასაკმარისია, რომ ეს მეთოდი სრულად დამტკიცდეს.
ეს მონაცემები ნათლად აჩვენებს, რომ TKOS განსაკუთრებით რეზისტენტულია ტრადიციული მკურნალობის მეთოდების მიმართ, და მოწოდებულია გადაუდებელი აუცილებლობა ახალი, ინოვაციური თერაპიების განვითარებისთვის, რათა ვუმკურნალოთ ამ მძიმე და პოტენციურად სასიკვდილო დაავადებას. პოტენციური თერაპიული მიდგომა შეიძლება იყოს ცილის ჩანაცვლებითი თერაპია გენური თერაპიის მეშვეობით. რადგან TKOS-ის პაციენტები ბუნებრივად არ გამოყოფენ ტრიადინს (ე.წ. “natural knockout”-ები არიან), TKOS იდეალურ კანდიდატად მიიჩნევა ცილის ჩანაცვლებითი მკურნალობისთვის, რომელშიც ნორმალური გულის ტრიადინის იზოფორმა გადაეცემა ადენოასოცირებული ვირუსის (AAV) მე-9 სეროტიპის მეშვეობით. ნორმალური ტრიადინის შეყვანამ TKOS-ის მქონე პაციენტის კარდიომიოციტებში შესაძლოა გაასწოროს არითმიული ფენოტიპი და თეორიულად გამოაჯანმრთელოს პაციენტი. სინამდვილეში, ამგვარი მიდგომა უკვე ნაჩვენებია, რომ ეფექტურია-რეცესიულად გადაცემადი CASQ2-ით გამოწვეული CPVT2-ის სამკურნალოდ, როგორც თაგვებში, ისე ადამიანის ინდუცირებულ პლურიპოტენტურ ღეროვან უჯრედებზე დამზადებულ კარდიომიოციტებში, WT-CASQ2 ცილის AAV9-ს მეშვეობით მიწოდებისას.
ამიტომ, იმისათვის, რომ უკეთ ვიზრუნოთ ამ მძიმე და მკურნალობაზე რეზისტენტული დაავადების მქონე პაციენტებზე, საჭიროა პაციენტების მუდმივ ჩართვა ITKOSR რეესტრში, რათა:გაგებულ იქნას TKOS-ის ბუნება და მოემზადოს კარგად დახასიათებული პაციენტთა ჯგუფი TRDN გენური თერაპიის მომავალი კლინიკური კვლევებისთვის.
ქვემოთ განხილულია ქეისები, რომლებსაც გულის უეცარი გაჩერება და ტრიადინის ნოკაუტის სინდრომის დიაგნოზი დაუსვეს
4.5 წლის ბიჭუნა მოთავსდა კლინიკაში ფიზიკური დატვირთვის დროს სინკოპეს გამო და ჩაუტარდა გულ-ფილტვის რეანიმაცია უეცარი გულის გაჩერების გამო. მას ჰქონდა უეცარი კარდიული სიკვდილის შესახებ ოჯახური ისტორია. ფიზიკური გამოკვლევა ნორმალური იყო, მაგრამ აღინიშნებოდა QTc ინტერვალის სასაზღვრო გახანგრძლივება. ადრენალინის პროვოკაციული ტესტის დროს დაფიქსირდა ორმხრივი არასტაბილური პოლიმორფული პარკუჭოვანი ტაქიკარდია. გენეტიკურ ანალიზში, TRDN გენში აღმოჩენილი იქნა c.568dupA, pII190Asnfs 2 ფრეიმშიფტის ვარიანტის ჰომოზიგოტური მუტაცია. ჩატარდა გულშიდა დეფიბრილატორის იმპლანტაცია. პროპრანოლოლისა და ფლეკაინიდის კომბინირებული მკურნალობით.
7.5 წლის ბიჭუნა კლინიკაში გადაიყვანეს მორეციდივე სინკოპეს და გულის გაჩერების ეპიზოდის ისტორიით. ოჯახურ ანამნეზში არ იყო უეცარი კარდიული სიკვდილის, სინკოპეს ან არითმიის შემთხვევები დაფიქსირებული. ფიზიკური გამოკვლევა, ელექტროკარდიოგრაფია, ექოკარდიოგრაფია და 24-საათიანი რიტმის ჰოლტერის მონიტორინგი ნორმალური იყო და ფიზიკური დატვირთვის ტესტის დროს აღმოჩენილი იქნა ორმხრივი პარკუჭოვანი ტაქიკარდია. გენეტიკურმა ანალიზმა გამოავლინა TRDN გენში ფრეიმშიფტის ვარიანტის ჰომოზიგოტური მუტაცია. ამ შემთხვევაშიც ჩატარდა გულშიდა დეფიბრილატორის იმპლანტაცია. პროპრანოლოლისა და ფლეკაინიდის კომბინირებული მკურნალობით. ორივე ქეისის შემთხვევაში მკურნალობა წარმატებული იყო და მსგავსი სიმპტომი არ დაფიქსირებულა.
2023 წლის 19 მარტს, კვლევის სუბიექტად შეირჩა ბავშვი, რომელიც 3 დღით ადრე გულის უეცარი გაჩერების გამო, მოათავსეს სიან ჯიაოტონგის უნივერსიტეტის ბავშვთა საავადმყოფოში. გენომური დნმ-ის ექსტრაქციისა და ეგზომის სეკვენირების (WES) მიზნით ბავშვისა და მისი მშობლებისგან აღებული იქნა პერიფერიული სისხლის ნიმუშები. პათოგენური ვარიანტების ოჯახური ვალიდაციის მიზნით ჩატარდა სენგერის სეკვენირება.
შედეგები: ბავშვს ფიზიკური დატვირთვის შემდეგ განუვითარდა სინკოპე და გულის გაჩერება. ელექტროკარდიოგრაფიულმა გამოკვლევამ გამოავლინა QTc ინტერვალის გახანგრძლივება, T-ტალღის ინვერსია პრეკორდიულ განხრებში V1-V3, პოლიმორფული პარკუჭოვანი ნაადრევი დარტყმა (VPB) და პარკუჭოვანი ტაქიკარდია (VT) გულისცემის მატებასთან ერთად. WES-ისა და სენგერის სეკვენირებამ აჩვენა, რომ ბავშვს ჰქონდა TRDN გენის ჰომოზიგოტური c.463del(p.E155Kfs*20) ვარიანტი, რომლის მიმართაც ორივე მშობელი ჰეტეროზიგოტური იყო. ბავშვს საბოლოოდ დაუსვეს TKOS დიაგნოზი.
დასკვნა:TRDN გენის ჰომოზიგოტური c.463del(p.E155Kfs20) ვარიანტი, სავარაუდოდ, ამ ბავშვში გულის გაჩერების პათოგენეზის საფუძველი იყო. ზემოაღნიშნულმა აღმოჩენამ განამტკიცა TRDN გენის მუტაციური სპექტრი.
თანდაყოლილი მიოპათია, რომელიც დაკავშირებულია ტრიადინის ნოკაუტის სინდრომთან
ამ კვლევის მიზანი იყო ტრიადინის ნოკაუტის სინდრომის დროს ჩონჩხის კუნთების ფენოტიპის ანალიზი. ტრიადინი გულისა და ჩონჩხის კუნთების კალციუმის გამოთავისუფლების კომპლექსის კომპონენტია. ჩატარდა კლინიკური შეფასება, გაანალიზდა მორფოლოგიური მახასიათებლები სინათლისა და ელექტრონული მიკროსკოპიის გამოყენებით და იმუნოლოკალიზებული ტრიადინი ჩონჩხის კუნთში.
კვლევა-6 წლის ბიჭს, რომელსაც მთელი ცხოვრების განმავლობაში კუნთების სისუსტე ჰქონდა, ჰქონდა ტრიადინის ნოკაუტის სინდრომი, რომელიც გამოწვეული იყო ტრიადინის რთული ჰეტეროზიგოტური ნულოვანი მუტაციებით. დელტოიდური კუნთის ნიმუშის სინათლის მიკროსკოპია აჩვენებს მრავალ მცირე პათოლოგიურ სივრცეს ყველა კუნთოვან ბოჭკოში. ელექტრონული მიკროსკოპია ავლენს სარკოპლაზმური ბადის გვერდითი ცისტერნების ფოკალურად განაწილებულ გაფართოებას და დეგენერაციას და ტრიადინის დაკარგვას შენახული გვერდითი ცისტერნებიდან.
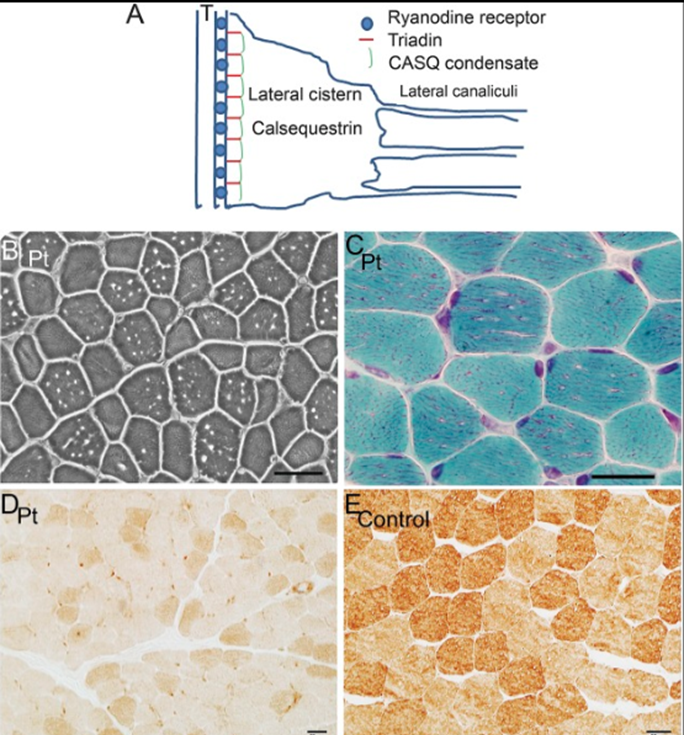
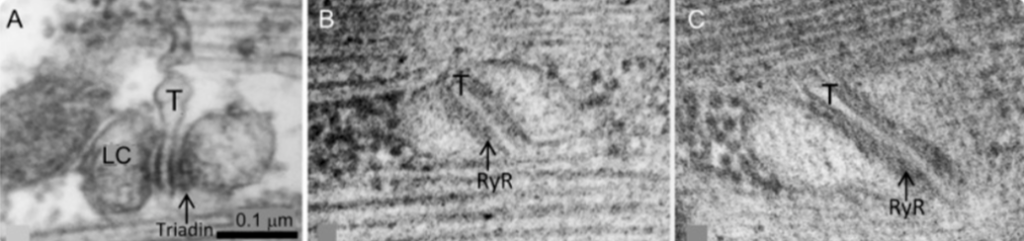
ადამიანებში ტრიადინის არარსებობამ შეიძლება გამოიწვიოს თანდაყოლილი მიოპათია, რომელიც დაკავშირებულია სარკოპლაზმური ბადის კომპონენტებში ღრმა პათოლოგიურ ცვლილებებთან. რატომ უვითარდებათ ჩონჩხის კუნთების ფენოტიპი მხოლოდ ზოგიერთ ტრიადინის დეფიციტურ პაციენტს, კვლავ გადაუჭრელი კითხვაა.
დასკვნები
მიუხედავად იმისა, რომ TKOS იშვიათი დაავადებაა და არ ფიქსირდება მოულოდნელი სიკვდილის ფართომასშტაბიან კვლევებში, ის მაინც რჩება მავნე და პოტენციურად სასიკვდილო დარღვევად, რომელიც შედარებადია სხვა იშვიათ დაავადებებთან, როგორიცაა კალმოდულინოპათია (calmodulinopathy) და ტიმოთეს სინდრომი (Timothy syndrome).
TKOS-ის მქონე პაციენტებს სიცოცხლისთვის საშიში გულ-სისხლძარღვთა მოვლენები ძალიან პატარა ასაკში უვლინდებათ, და თითქმის 75%-მა განიცადა მძიმე კარდიული ეპიზოდები მიუხედავად ჩვეულებრივი მკურნალობის მიღებისა.
ამიტომ, დაავადების სიმძიმე და მკურნალობაზე რეზისტენტულობა მოსალოდნელად იმეორებს მკვეთრ აუცილებლობას, რომ ჩატარდეს დამატებითი კვლევები და განვითარდეს ახალი, ეფექტური თერაპიული მეთოდები ამ პაციენტების უკეთ მოვლის მიზნით.
References
BRAUNWALD’S HEART DISEASE, 12th edition, chapter 63, Genetics of Cardiac Arrhythmias, Pg.1195-1196
A very rare cause of sudden cardiac arrest in children: triadin knockout syndrome(https://pubmed.ncbi.nlm.nih.gov/35481495/)
[Clinical and genetic analysis of a case of Triadin knockout syndrome due to variant of TRDN gene and a literature review] (https://pubmed.ncbi.nlm.nih.gov/35481495/)
International Triadin Knockout Syndrome Registry: The Clinical Phenotype and Treatment Outcomes of Patients With Triadin Knockout Syndrome (https://www.ahajournals.org/doi/10.1161/CIRCGEN.118.002419 )
Triadin Knockout Syndrome Is Absent in a Multi-Center Molecular Autopsy Cohort of Sudden Infant Death Syndrome and Sudden Unexplained Death in the Young and Is Extremely Rare in the General Population (https://www.ahajournals.org/doi/10.1161/CIRCGEN.119.002731 )
Congenital myopathy associated with the triadin knockout syndrome (https://pubmed.ncbi.nlm.nih.gov/28202702/)